Los pacientes con diabetes tienen un riesgo incrementado de sufrir enfermedad cardiovascular, debido a las características de esta patología. Cualquier fármaco utilizado para el tratamiento de esta enfermedad crónica debe tener en cuenta este aspecto, y ya desde hace unos años se requiere que al menos no incrementen ese riesgo a largo plazo.
En 2008 y 2012 se publicaron las guías de la Food and Drug Administration (FDA)1 y de la Agencia Europea de Medicamentos2,3 sobre investigación clínica de medicamentos para el tratamiento de la diabetes mellitus. Estos documentos incluyeron un apartado con requisitos que debían cumplir estos fármacos en cuanto a seguridad cardiovascular, y cómo llevar a cabo esa evaluación.
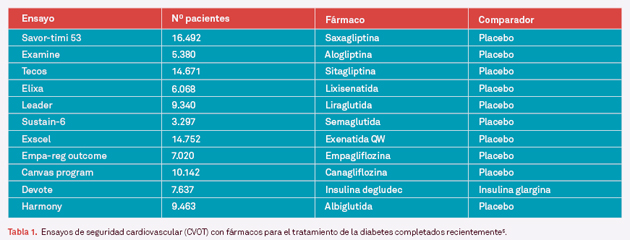
Fundamentalmente, un nuevo fármaco debe confirmar que no induce en los pacientes un mayor riesgo de producir enfermedad cardiovascular (ECV) que un placebo o un fármaco que ya haya demostrado este hecho. Si un metaanálisis de datos de estudios en fase 2 y 3 no puede confirmar este aspecto, se debe realizar un ensayo clínico específico (CVOT, Cardiovascular Outcome Trial). Para ello, se deben evaluar variables principales como mortalidad por enfermedad cardiovascular, infarto de miocardio no fatal o evento cerebrovascular no fatal (3-point MACE o Major Adverse Cardiovascular Events)4,5. Al necesitarse un elevado número de estos eventos en las dos ramas del estudio para poder obtener resultados significativos, se requiere la inclusión de un gran número de pacientes durante un largo tiempo (generalmente de 3 a 5 años). En la tabla 1 se muestra una lista de algunos de estos ensayos y sus características.
La realización de estos ensayos plantea una serie de retos de todo tipo, desde operativos (cómo llevarlos a cabo) hasta de diseño de los mismos (elección de variables primarias y secundarias, etc.). Dada la experiencia de nuestro departamento de investigación clínica en este tipo de estudios (de los que hemos realizado ya cuatro), quisiera exponer alguno de esos importantes retos.
En cuanto al diseño de los mismos. se plantean varios debates sobre:
• población del estudio: generalmente debe incluirse una población parecida a la que se pretende tratar con el fármaco una vez autorizado, pero dado que se necesita un número de eventos cardiovasculares elevado, se permite la inclusión de un porcentaje más elevado de pacientes con alto riesgo de esta enfermedad CV (pacientes ya con historia previa de infarto, ictus, etc.).
Esto plantea algunas diferencias entre estudios realizados con distintos productos y poblaciones diferentes, que debe describirse y evaluarse bien a la hora de interpretar los resultados. Deberían distinguirse los resultados entre la población con historia CV previa y la población que no.
• “endpoints” a evaluar: hay bastante consenso en que los tres principales “endpoints” que componen la variable principal son los adecuados (mortalidad, infarto de miocardio no fatal y evento cerebrovascular no fatal), pero se plantea el poder consensuar otros secundarios como pueden ser el tiempo hasta la primera hospitalización por un evento CV o la progresión de la retinopatía diabética. También se plantea incluir en estos ensayos posibles variables que estudian los efectos microvasculares, como nefropatía diabética. Un mayor debate es necesario para estandarizar el uso de estas variables secundarias.
• uso de placebo como grupo control: en el caso de que no existan fármacos del mismo grupo con seguridad CV bien caracterizada, el grupo comparador puede ser placebo, si es éticamente aceptable. Su uso es controvertido, dada la duración de estos estudios, aunque realmente a los pacientes se les puede intensificar el tratamiento si es necesario con fármacos comercializados permitidos en el protocolo del estudio. Existe un debate sobre si el comparador debería ser de forma estándar un fármaco con un beneficio CV ya probado. En este caso, conseguir un beneficio CV extra sería más difícil que frente a placebo, pero un porcentaje mayor de pacientes se beneficiarían del tratamiento con estos efectos.
Estos ensayos clínicos también plantean dificultades operativas y especificidades propias en su ejecución, como son:
• número de pacientes en el estudio: como se ha comentado anteriormente, se requiere un número muy elevado de pacientes en estos ensayos, y la duración de los mismos es muy larga. Esto requiere para las compañías que realizamos estos proyectos una inversión en recursos elevada. Los protocolos de los ensayos son de baja complejidad generalmente, lo que lleva a tratar de incluir un elevado número de pacientes por centro para maximizar su ejecución. Usualmente participan muchos países y la esto también complica la logística del estudio.
• selección de los centros con los especialistas adecuados: los pacientes que se incluyen en estos ensayos son llevados por diversos especialistas, que incluyen a endocrinólogos, cardiólogos, neurólogos, médicos de atención primaria o medicina interna. Dada la importancia que tiene en estos estudios la retención del paciente durante toda la duración del estudio (ver siguiente punto), se hace necesario identificar caso a caso cuál de ellos es el que tendrá una relación más directa con él. La colaboración entre servicios es esencial en muchos casos y eso añade una complejidad extra en estos estudios en cuanto a la coordinación y ejecución de las tareas propias de un ensayo clínico. Dado que ya se han realizado muchos de estos estudios, ya se han establecido estas redes de especialistas y no es tan complicado el encontrar buenos centros para la realización de estos proyectos.
• retención de los pacientes en el estudio: dado que conseguir el número de eventos CV predeterminado es el objetivo primario de estos estudios, se hace necesario un seguimiento de todos los pacientes que hayan sido expuestos al fármaco para poder “capturar” dichos eventos una vez que se produzcan. Desde el punto de vista ético, un paciente puede abandonar un ensayo clínico en cualquier momento sin tener que dar explicaciones. Sin embargo, en la totalidad de estos estudios se fija como objetivo operativo una tasa de abandono de un máximo de un 1%. Este límite es crucial para poder conseguir unos resultados significativos en el estudio. Generalmente se utilizan diversas estrategias para conseguir que el paciente quiera completar el estudio. como son:
* realización por parte de los equipos investigadores de actividades de concienciación sobre la patología y la importancia de estos tratamientos
* menor número de procedimientos en el estudio que en otros ensayos, con menos visitas al centro y más contactos telefónicos
* posibilidad de que incluso el paciente deje el tratamiento del estudio sin que abandone el mismo. El tiempo de exposición al fármaco es menor, pero se asegura que, si ocurre un evento, esta información se pueda recoger
* en algunos casos, solamente se recoge información al final del estudio sobre el estado vital del paciente, sin que éste tenga que atender a las visitas programadas en el estudio
• clasificación de los eventos CV: dada la necesidad de verificar los eventos que se van recogiendo durante el estudio, se hace necesaria una evaluación centralizada caso a caso. Para ello, se recoge información de cada evento y se evalúa por un comité que “adjudica” o clasifica el evento según estándares del protocolo. Esto añade un punto más de complejidad a estos ensayos. pero asegura el objetivo principal del mismo.
Como resumen de este artículo, podemos recalcar que los ensayos de seguridad cardiovascular de fármacos para la diabetes son una parte muy importante del desarrollo clínico de estos medicamentos, no sólo por su dimensión (tiempo y volumen de datos necesarios), sino también por que conducen a perfilar los posibles beneficios de estas moléculas en los pacientes diabéticos.
Existe un debate en cómo deben llevarse a cabo estos proyectos, y de esta forma las guías de la FDA y EMA están en revisión para asegurar que se obtienen los resultados necesarios para establecer el perfil cardiovascular de forma adecuada y rigurosa.
En los próximos años se tendrán también que tener en cuenta otros temas como puede ser la utilización de datos de vida real o como incorporar otros parámetros CV para establecer ese perfil de seguridad cardiovascular.
Referencias
1. Food and Drug Administration. Guidance for industry diabetes mellitus—evaluating cardiovascular risk in new antidiabetic therapies to treat type 2 diabetes. Washington. D.C.: US Department of Health and Human Services; 2008.
2. European Medicines Agency. Guideline on clinical investigation of medicinal products in the treatment or prevention of diabetes mellitus. 2012. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500129256.pdf. (Accessed 26 February 2015).
3. European Medicines Agency. Reflection paper on assessment of cardiovascular risk of medicinal products for the treatment of cardiovascular and metabolic diseases. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2016/03/WC500203804.pdf
(18 April 2017).
4. Schnell O. Standl E. Catrinoiu D. Genovese S. Lalic N. Lalic K. Skrha J. Valensi P y Ceriello A. Report from the 3rd Cardiovascular Outcome Trial (CVOT) Summit of the Diabetes & Cardiovascular Disease (D&CVD) EASD Study Group. Cardiovasc Diabetol. 2018;17:30
5. Cefalu WT, Kaul S, Gerstein HC, et al. Cardiovascular outcomes trials in type 2 diabetes: where do we go from here? Reflections from a Diabetes Care Editors’ Expert Forum. Diabetes Care 2018;41:14–31











